 DAA既治療(国内)
DAA既治療(国内)
本剤の「警告・禁忌を含む使用上の注意」等については
注意事項等情報(電子化された添付文書)をご参照ください。
DAAによる治療歴を有する患者を対象とした国内第3相臨床試験(エプクルーサ配合錠+RBV 24週間投与群)
エプクルーサ配合錠を1日1回1錠、リバビリン併用下で24週間服用することにより、DAA治療歴のあるC型慢性肝炎、C型代償性肝硬変患者さんの96.7%がSVR12を達成しました。また、60例中21例(35.0%)に副作用が認められました。
試験概要
| 目的 | 直接作用型抗ウイルス薬(DAA)による前治療が不成功であったジェノタイプ1又は2のC型慢性肝炎又はC型代償性肝硬変の患者を対象に、エプクルーサ配合錠+リバビリン(RBV)12週間又は24週間投与の有効性と安全性を検討する。 |
|---|---|
| 対象 | DAAによる前治療が不成功であったジェノタイプ1又は2のC型慢性肝炎又はC型代償性肝硬変患者117例。 (肝硬変の判定基準には、肝生検又はFibroscan®の結果(>12.5kPa)もしくはFibroTestスコア(≧0.75)を用いた) |
| 試験デザイン | 多施設共同、無作為化、非盲検、並行群間比較試験。 スクリーニング時の肝硬変の有無及びHCVジェノタイプ(ジェノタイプ1又は2)による層別ランダム化を行った。約20例は、スクリーニング時にChild-Pugh(CP)分類Aの代償性肝硬変を有する被験者を登録することとした。約90例はジェノタイプ1、約20例はジェノタイプ2のHCV感染被験者が登録されるよう計画した。 |
| 方法 | 対象をエプクルーサ配合錠+RBV12週間投与群※又はエプクルーサ配合錠+RBV24週間投与群に無作為に割り付け、経口投与を行った[エプクルーサ配合錠(ソホスブビル/ベルパタスビル:400mg/100mg)は1日1回、RBVは600〜1,000mg(体重に基づいた1日用量)を分割投与]。 |
| 主要評価項目 | SVR12率[投与終了から12週間後のHCV RNA量が定量下限(LLOQ)
|
| 副次評価項目 | SVR4率、SVR24率、ウイルス学的転帰及び試験治療下の来院時ごとのウイルス陰性化率¶、投与終了までのHCVRNAの絶対量及びベースラインからの変化量、治療中及び治療後の耐性関連変異の特性。 |
| その他の有効性評価項目 | ベースラインのNS5A及びNS5B耐性関連変異の有無別のサブグループ解析。 |
| 解析計画 | 主要評価項目は、Full Analysis Set(FAS)においてClopper-Pearson法に基づく点推定値及び正確な両側95%信頼区間(CI)をそれぞれの投与群に対して算出した。2つの投与群それぞれのジェノタイプ1のHCV感染被験者のSVR12率を、ヒストリカルコントロールのSVR12率50%と、有意水準を0.025とする両側正確1標本二項検定を用いて比較した。ジェノタイプ2のHCV感染被験者においては、統計的仮説検定は実施しなかった。背景因子別のサブグループ解析は、年齢(65歳以上/未満)、肝硬変の有無、HCVジェノタイプ/サブタイプ、前治療のDAA(NS5A + NS5B / NS5A + NS3/4A / NS5A + NS5B + NS3/4A / NS5Bのみ / NS5B+NS3/4A)、ベースライン時のNS5A及びNS5B耐性関連変異の有無等について実施することにした。 |
SVR:
sustained virologic response(持続的ウイルス陰性化)
†
定量下限(LLOQ):本試験のHCV RNA量の定量にはCOBAS® AmpliPrep /COBAS® TaqMan® HCV Quantitative Test
v2.0を用い、本分析法の定量下限(LLOQ)は15IU/mLでした。
¶
HCV RNA<LLOQを達成した患者割合。
◆ DAAによる治療歴を有する患者を対象とした国内第3相臨床試験:試験デザイン
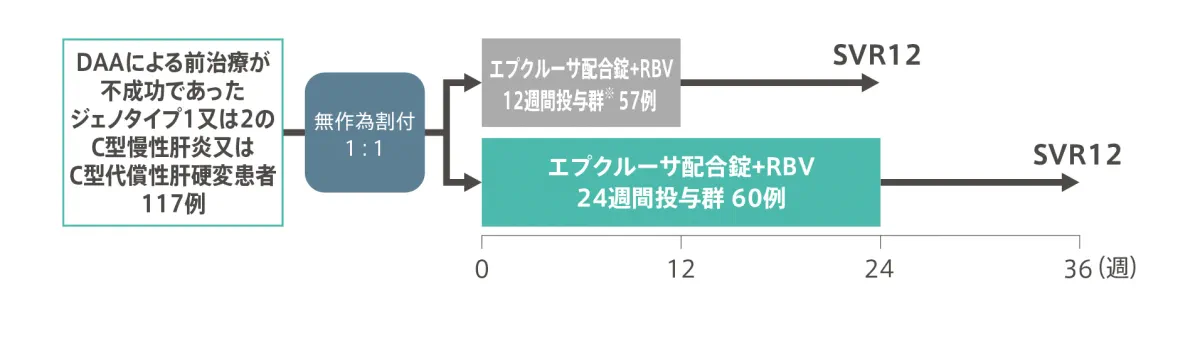
※ DAAによる治療歴を有するC型慢性肝炎又はC型代償性肝硬変に対する12週間投与は国内未承認。以降は、承認を受けた用法・用量の成績のみを紹介しています。
社内資料:承認時評価資料(国内第3相臨床試験:GS-US-342-3921)
Izumi N, et al. Hepatol Int. 12(4): 356-367, 2018
利益相反: 本研究はギリアド・サイエンシズ, Inc.の資金提供および支援により行われた。本論文の著者には、ギリアド・サイエンシズ, Inc.の社員が含まれる。本論文の著者には、ギリアド・サイエンシズ,
Inc.より講演料等を受領した者が含まれる。
【用法・用量】(抜粋)
〈前治療歴を有するC型慢性肝炎又はC型代償性肝硬変におけるウイルス血症の改善〉
リバビリンとの併用において、通常、成人には、1日1回1錠(ソホスブビルとして400mg及びベルパタスビルとして100mg)を24週間経口投与する。
リバビリンの併用に際しては、リバビリンの注意事項等情報(電子化された添付文書)を参照すること。
患者背景
患者背景
| エプクルーサ配合錠+RBV24週間投与群 (n=60) |
|||
|---|---|---|---|
| 年齢中央値(範囲) | 64歳(35-79) | ||
| ≧65歳 | 29例(48.3%) | ||
| 男性 | 27例(45.0%) | ||
| BMI(平均値±標準偏差),kg/m2 | 23.5±3.70 | ||
| IL28B CC 遺伝子型※1 | 27例(45.0%) | ||
| HCV RNA 量(平均値±標準偏差), (範囲), log10 IU/mL |
6.2±0.58(4.3-7.1) | ||
| HCV RNA ≧ 5 log10 IU/mL | 57例(95.0%) | ||
| ALT値中央値(Q1※2〜Q3※3), U/L | 33(18〜58) | ||
| ジェノタイプ | 1a | 1例(1.7%) | |
| 1b | 47例(78.3%) | ||
| 2(未確定) | 4例(6.7%) | ||
| 2a/2c | 4例(6.7%) | ||
| 2b | 4例(6.7%) | ||
| 代償性肝硬変 | 21例(35.0%) | ||
| 前治療のDAA薬剤数 | 1剤 | 8例(13.3%) | |
| 2剤 | 41例(68.3%) | ||
| 3剤以上 | 11例(18.3%) | ||
- IL28B領域の一塩基多型(SNPs)の解析にあたり、本試験においては、rs12979860の解析によりSNPsのメジャーアリル(CC)もしくはマイナーアリル(CT、TT)を同定している。
- 25パーセンタイル
- 75パーセンタイル
【DAAを含む前治療の内訳※】
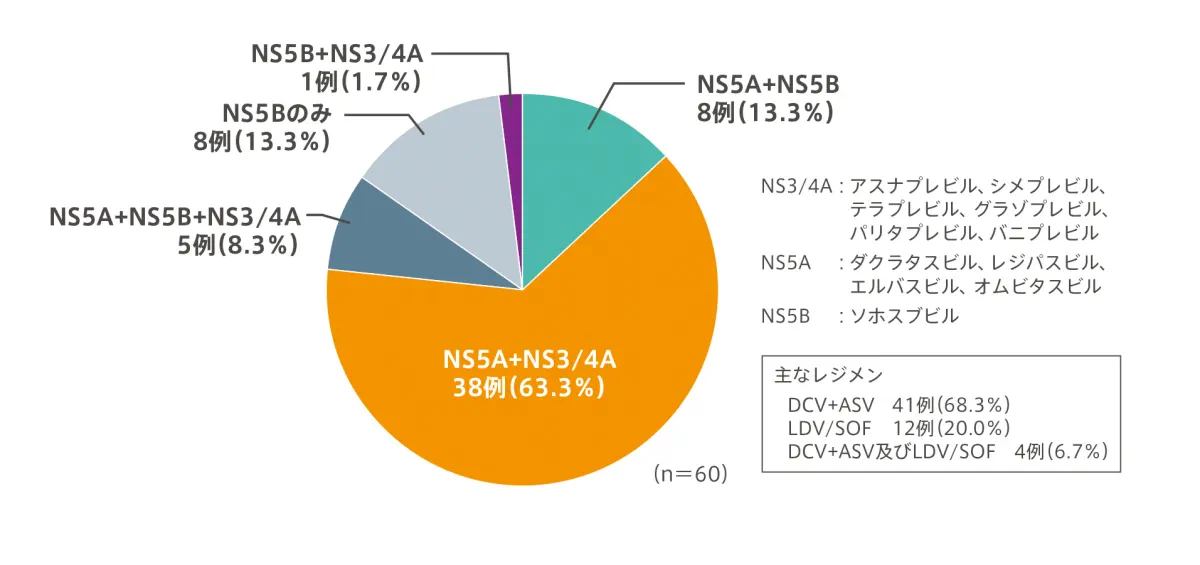
- 複数回の治療歴がある場合は薬剤系統ごとの、のべ集計
◆ ベースラインの耐性関連変異
| エプクルーサ配合錠+RBV24週間投与群 | ||||
|---|---|---|---|---|
| 全体 (n=60) |
GT1 (n=48) |
GT2 (n=12) |
||
| NS5A 耐性関連変異 |
L31 変異 ± 他の変異 | 52例 (86.7%) |
42例 (87.5%) |
10例 (83.3%) |
| P32欠損 ± 他の変異 | 3例 (5.0%) |
3例 (6.3%) |
0例 (0%) |
|
| Y93 変異 ± 他の変異 | 39例 (65.0%) |
39例 (81.3%) |
0例 (0%) |
|
エプクルーサ配合錠+RBV24週間投与群において核酸型NS5B阻害剤に対する耐性関連変異はL159F(2例)、M289I(1例)、V321I(1例)でした。
耐性関連変異は、下記のアミノ酸部位に対応する標準配列からの変異をディープシークエンス法を用いて検出しました。
| 耐性関連変異 | ジェノタイプ | 検討したアミノ酸部位 |
|---|---|---|
| NS5A | GT1a | 24、26、28、30、31、32、38、58、92又は93 |
| GT1b | 28、31、32、58、92又は93 | |
| GT2a、GT2b | 24、28、30、31、32、38、58、92又は93 | |
| NS5B | ー | 96、142、159、237、282、289、320又は321 |
社内資料:承認時評価資料(国内第3相臨床試験:GS-US-342-3921)
Izumi N, et al. Hepatol Int. 12(4): 356-367, 2018
利益相反: 本研究はギリアド・サイエンシズ, Inc.の資金提供および支援により行われた。本論文の著者には、ギリアド・サイエンシズ, Inc.の社員が含まれる。本論文の著者には、ギリアド・サイエンシズ,
Inc.より講演料等を受領した者が含まれる。
SVR12率(主要評価項目、サブグループ解析)
エプクルーサ配合錠+RBV24週間投与により、患者全体の96.7%(58/60例)がSVR12[投与終了12週間後の持続的ウイルス陰性化]を達成しました(主要評価項目)。ジェノタイプ別SVR12率は、ジェノタイプ1が97.9%(47/48例)、ジェノタイプ2が91.7%(11/12例)でした(サブグループ解析)。
その他の背景因子別(年齢、代償性肝硬変の有無)及び前治療のDAA別のSVR12率は以下のとおりです。なお、本試験では、96.7%(58/60例)の患者が本剤の投与を完遂しました。
◆ SVR12率※(主要評価項目)

- 投与終了12週間後の持続的ウイルス陰性化率
◆ ジェノタイプ別SVR12率(サブグループ解析)

社内資料:承認時評価資料(国内第3相臨床試験:GS-US-342-3921)
Izumi N, et al. Hepatol Int. 12(4): 356-367, 2018
利益相反: 本研究はギリアド・サイエンシズ, Inc.の資金提供および支援により行われた。本論文の著者には、ギリアド・サイエンシズ, Inc.の社員が含まれる。本論文の著者には、ギリアド・サイエンシズ,
Inc.より講演料等を受領した者が含まれる。
背景因子別SVR12率(サブグループ解析)
背景因子別SVR12率は以下の通りです。
◆年齢別

◆代償性肝硬変有無別

【特定の背景を有する患者に関する注意】(抜粋)
9.8 高齢者
患者の状態を観察しながら慎重に投与すること。一般に生理機能が低下しており、既往歴や合併症を伴っていることが多くみられる。
◆ 前治療のDAA別SVR12率
| 前治療のDAA | 全体 (n=60) |
GT1 (n=48) |
GT2 (n=12) |
|---|---|---|---|
| NS5A+NS5B | 8/8 | 7/7 | 1/1 |
| NS5A+NS3/4A | 37/38 (97.4%) |
36/37 (97.3%) |
1/1 |
| NS5A+NS5B+NS3/4A | 5/5 | 4/4 | 1/1 |
| NS5Bのみ | 8/8 | ー | 8/8 |
| NS5B+NS3/4A | 0/1 | ー | 0/1 |
NS5A:ダクラタスビル、レジパスビル、エルバスビル、オムビタスビル
NS5B:ソホスブビル
NS3/4A:アスナプレビル、シメプレビル、テラプレビル、グラゾプレビル、パリタプレビル、バニプレビル
社内資料:承認時評価資料(国内第3相臨床試験:GS-US-342-3921)
Izumi N, et al. Hepatol Int. 12(4): 356-367, 2018
利益相反: 本研究はギリアド・サイエンシズ, Inc.の資金提供および支援により行われた。本論文の著者には、ギリアド・サイエンシズ, Inc.の社員が含まれる。本論文の著者には、ギリアド・サイエンシズ,Inc.より講演料等を受領した者が含まれる。
ベースラインのNS5A及びNS5B耐性関連変異の有無別SVR12率(その他の有効性評価項目)
ベースライン時に患者の93.3%(56/60例)でNS5A耐性関連変異が認められました。エプクルーサ配合錠+RBV24週間投与により、NS5A耐性関連変異が検出された患者の96.4%(54/56例)がSVR12を達成し、検出されなかった患者では4例中4例がSVR12を達成しました。NS5A耐性変異別のSVR12率は以下のとおりです。
また、ベースライン時に核酸型NS5B阻害剤に対する耐性関連変異が検出された患者4例は、全員がSVR12を達成しました。
◆ ベースラインのNS5A耐性関連変異の有無別(15%カットオフ)SVR12率

社内資料:承認時評価資料(国内第3相臨床試験:GS-US-342-3921)
Izumi N, et al. Hepatol Int. 12(4): 356-367, 2018
利益相反: 本研究はギリアド・サイエンシズ, Inc.の資金提供および支援により行われた。本論文の著者には、ギリアド・サイエンシズ, Inc.の社員が含まれる。本論文の著者には、ギリアド・サイエンシズ,
Inc.より講演料等を受領した者が含まれる。
◆NS5A耐性関連変異別SVR12率(サブグループ解析)
| 全体 | GT1 | GT2 | ||
|---|---|---|---|---|
| NS5A 耐性関連変異 |
L31 変異 ± 他の変異 (n=52) |
50/52 (96.2%) |
41/42 (97.6%) |
9/10 (90.0%) |
| P32欠損 ± 他の変異 (n=3) |
2/3 | 2/3 | − | |
| Y93 変異 ± 他の変異 (n=39) |
39/39 (100%) | 39/39 (100%) | − | |
耐性関連変異は、下記のアミノ酸部位に対応する標準配列からの変異をディープシークエンス法を用いて検出しました。
| 耐性関連変異 | ジェノタイプ | 検討したアミノ酸部位 |
|---|---|---|
| NS5A | GT1a | 24、26、28、30、31、32、38、58、92又は93 |
| GT1b | 28、31、32、58、92又は93 | |
| GT2a、GT2b | 24、28、30、31、32、38、58、92又は93 | |
| NS5B | ー | 96、142、159、237、282、289、320又は321 |
社内資料:承認時評価資料(国内第3相臨床試験:GS-US-342-3921)
Izumi N, et al. Hepatol Int. 12(4): 356-367, 2018
利益相反: 本研究はギリアド・サイエンシズ, Inc.の資金提供および支援により行われた。本論文の著者には、ギリアド・サイエンシズ, Inc.の社員が含まれる。本論文の著者には、ギリアド・サイエンシズ,
Inc.より講演料等を受領した者が含まれる。
SVR24率(副次評価項目)
エプクルーサ配合錠+RBV 24週間投与によりSVR12を達成した全例がSVR24を達成しました。
ウイルス学的転帰(副次評価項目)
エプクルーサ配合錠+RBV24週間投与において、投与期間中のウイルス学的治療不成功は認められず、再燃は2例でした。
◆ ウイルス学的転帰
| 全体 (n=60) |
GT1 (n=48) |
GT2 (n=12) |
|
|---|---|---|---|
| SVR12 | 58/60(96.7%) | 47/48(97.9%) | 11/12(91.7%) |
| ウイルス学的治療不成功 | 2/60(3.3%) | 1/48(2.1%) | 1/12(8.3%) |
| 再燃例 | 2/60(3.3%) | 1/48(2.1%) | 1/12(8.3%) |
| 試験完了 | 2/58(3.4%) | 1/46(2.2%) | 1/12(8.3%) |
| 投与中止 | 0/2 | 0/2 | ー |
| 投与期間中のウイルス学的 治療不成功 |
0/60 | 0/48 | 0/12 |
| その他 | 0/60 | 0/48 | 0/12 |
社内資料:承認時評価資料(国内第3相臨床試験:GS-US-342-3921)
Izumi N, et al. Hepatol Int. 12(4): 356-367, 2018
利益相反: 本研究はギリアド・サイエンシズ, Inc.の資金提供および支援により行われた。本論文の著者には、ギリアド・サイエンシズ, Inc.の社員が含まれる。本論文の著者には、ギリアド・サイエンシズ,
Inc.より講演料等を受領した者が含まれる。
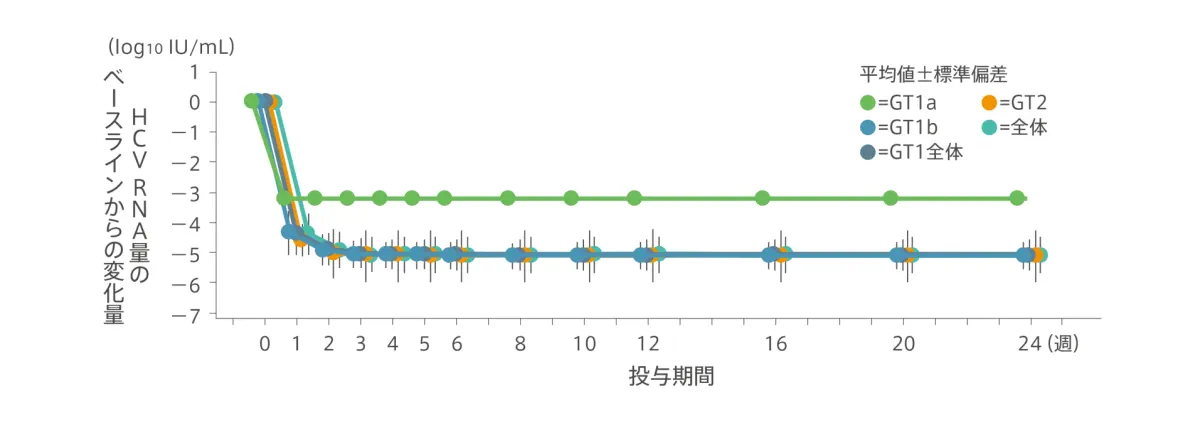
HCV RNA量の推移/ウイルス陰性化率(副次評価項目)
HCV RNA量は急速に減少し、投与開始後1週時点におけるHCV RNA量のベースラインからの変化量(平均値±標準偏差)は、全体で−4.36±0.698log10IU/mLでした。投与4週時点で98.3%(59/60例)がHCV RNA量LLOQ未満となり、投与8週時点から投与終了(24週)まで100%(8週:60/60例、12週:59/59例、24週:58/58例)がHCV RNA量LLOQ未満を維持しました。
◆ HCV RNA量のベースラインからの変化量

◆ ウイルス陰性化率※(%)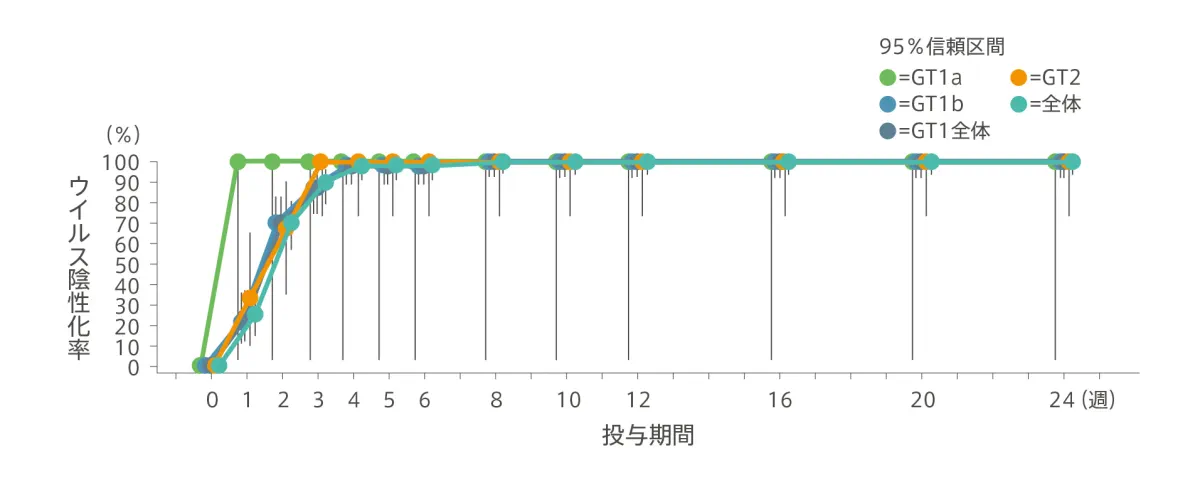
| 投与期間 | 0週間 | 1週間 | 2週間 | 3週間 | 4週間 | 5週間 | 6週間 | 8週間 | 12週間 | 24週間 |
|---|---|---|---|---|---|---|---|---|---|---|
| GT1 (n=48) |
0% (0/48) |
22.9% (11/48) |
70.8% (34/48) |
87.5% (42/48) |
97.9% (47/48) |
97.9% (47/48) |
97.9% (47/48) |
100% (48/48) |
100% (47/47) |
100% (46/46) |
| GT2 (n=12) |
0% (0/12) |
33.3% (4/12) |
66.7% (8/12) |
100% (12/12) |
100% (12/12) |
100% (12/12) |
100% (12/12) |
100% (12/12) |
100% (12/12) |
100% (12/12) |
| 全体 (n=60) |
0% (0/60) |
25.0% (15/60) |
70.0% (42/60) |
90.0% (54/60) |
98.3% (59/60) |
98.3% (59/60) |
98.3% (59/60) |
100% (60/60) |
100% (59/59) |
100% (58/58) |
※ HCV RNA量 < LLOQ(15 IU/mL)
社内資料:承認時評価資料(国内第3相臨床試験:GS-US-342-3921)
Izumi N, et al. Hepatol Int. 12(4): 356-367, 2018
利益相反: 本研究はギリアド・サイエンシズ, Inc.の資金提供および支援により行われた。本論文の著者には、ギリアド・サイエンシズ, Inc.の社員が含まれる。本論文の著者には、ギリアド・サイエンシズ,
Inc.より講演料等を受領した者が含まれる。
治療中及び治療後の耐性関連変異の特性(副次評価項目)
エプクルーサ配合錠+RBV24週間投与において、ウイルス学的治療不成功は60例中2例(3.3%)で、いずれも再燃でした。再燃した2例について、ベースライン時及び再燃が認められた時点で、NS5A及びNS5B領域のディープシークエンス解析を実施したところ、NS5A耐性関連変異の新たな出現は認められず、核酸型NS5B阻害剤に対する耐性関連変異は検出されませんでした。
◆ 再燃例においてベースライン時及び投与終了後に検出されたNS5A耐性関連変異(15%カットオフ)
| 投与 | ジェノタイプ | 肝炎/ 代償性肝硬変 |
前治療 | NS5A耐性関連変異 | ||
|---|---|---|---|---|---|---|
| ベースライン | 再燃時 | |||||
| 患者A | 完遂 | 1b | 肝炎 | DCV+ASV | L31I/V P32欠損 |
P32欠損 |
| 患者B | 完遂 | 2b | 肝炎 | SOF+RBV | L31M | L31M |
DCV:ダクラタスビル ASV:アスナプレビル SOF:ソホスブビル RBV:リバビリン
社内資料:承認時評価資料(国内第3相臨床試験:GS-US-342-3921)
Izumi N, et al. Hepatol Int. 12(4): 356-367, 2018
利益相反: 本研究はギリアド・サイエンシズ, Inc.の資金提供および支援により行われた。本論文の著者には、ギリアド・サイエンシズ, Inc.の社員が含まれる。本論文の著者には、ギリアド・サイエンシズ,
Inc.より講演料等を受領した者が含まれる。
安全性(主要評価項目)
【副作用発現状況】
前治療歴を有するC型慢性肝炎又はC型代償性肝硬変患者を対象に、エプクルーサ配合錠とRBVを24週間併用投与した国内第3相臨床試験において、60例中21例(35.0%)に副作用が認められました。
主な副作用は、貧血13例(21.7%)、倦怠感3例(5.0%)、そう痒症2例(3.3%)等でした。(承認時)
エプクルーサ配合錠+RBV24週間投与群において、死亡例及び重篤な副作用は認められませんでした。重篤な有害事象として、肝細胞癌2例、肝血管肉腫1例、肺炎1例が認められました。投与中止に至った有害事象として、うつ病1例、肝血管肉腫1例が認められました。
【ヘモグロビン値】
エプクルーサ配合錠+RBV24週間投与において、投与開始後に1回以上ヘモグロビン値10g/dL未満となった患者は60例中5例(8.3%)で、65歳未満が31例中2例(6.5%)、65歳以上が29例中3例(10.3%)でした。このうち65歳以上の1例で、投与開始後のヘモグロビン値が8.5g/dL未満となりました。本試験で輸血又はエリスロポエチン製剤を使用した患者はいませんでした。
なお、RBVの用量変更及び投与中止は、RBVの注意事項等情報(電子化された添付文書)に従いました。
◆ 投与開始後にヘモグロビン値が10又は8.5g/dL未満まで低下した患者の割合
| 全体 (n=60) |
65歳未満 (n=31) |
65歳以上 (n=29) |
|
|---|---|---|---|
| 投与開始後に1回以上ヘモグロビン値が 10g/dL未満となった患者数(%) |
5例(8.3%) | 2例(6.5%) | 3例(10.3%) |
| 投与開始後に1回以上ヘモグロビン値が 8.5g/dL未満となった患者数(%) |
1例(1.7%) | 0例 | 1例(3.4%) |
社内資料:承認時評価資料(国内第3相臨床試験:GS-US-342-3921)
Izumi N, et al. Hepatol Int. 12(4): 356-367, 2018
利益相反: 本研究はギリアド・サイエンシズ, Inc.の資金提供および支援により行われた。本論文の著者には、ギリアド・サイエンシズ, Inc.の社員が含まれる。本論文の著者には、ギリアド・サイエンシズ,
Inc.より講演料等を受領した者が含まれる。
◆ ヘモグロビン(中央値)の推移
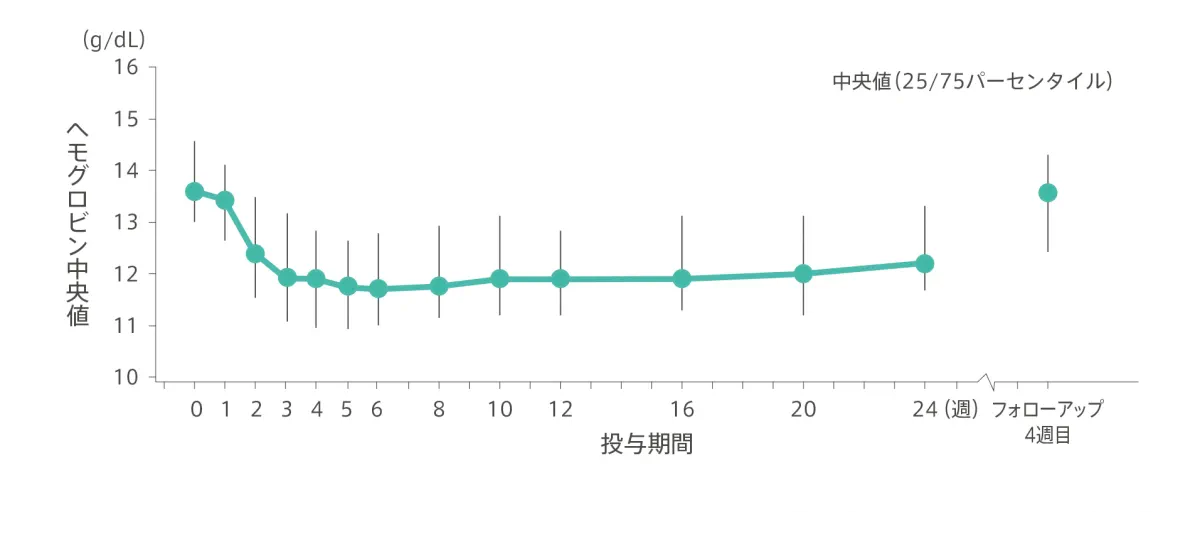
社内資料:承認時評価資料(国内第3相臨床試験:GS-US-342-3921)
Izumi N, et al. Hepatol Int. 12(4): 356-367, 2018
利益相反: 本研究はギリアド・サイエンシズ, Inc.の資金提供および支援により行われた。本論文の著者には、ギリアド・サイエンシズ, Inc.の社員が含まれる。本論文の著者には、ギリアド・サイエンシズ,
Inc.より講演料等を受領した者が含まれる。
〈重要な基本的注意〉(抜粋)
〈リバビリンとの併用の場合〉
8.5 投与開始前にヘモグロビン量が12g/dL以上であることを確認すること。貧血があらわれることがあるので、ヘモグロビン量を定期的に測定するなど観察を十分に行うこと。
また、投与中にリバビリンの用量調節や投与中止を必要とする副作用が発現した場合には、リバビリンの注意事項等情報(電子化された添付文書)を参照すること。
【ALT値の推移】
ALT(中央値)は、ベースライン時33U/L、2~8週時17U/L、10~12週時19U/L、24週時18U/L、フォローアップ4週目18U/Lでした。
◆ ALT(中央値)の推移
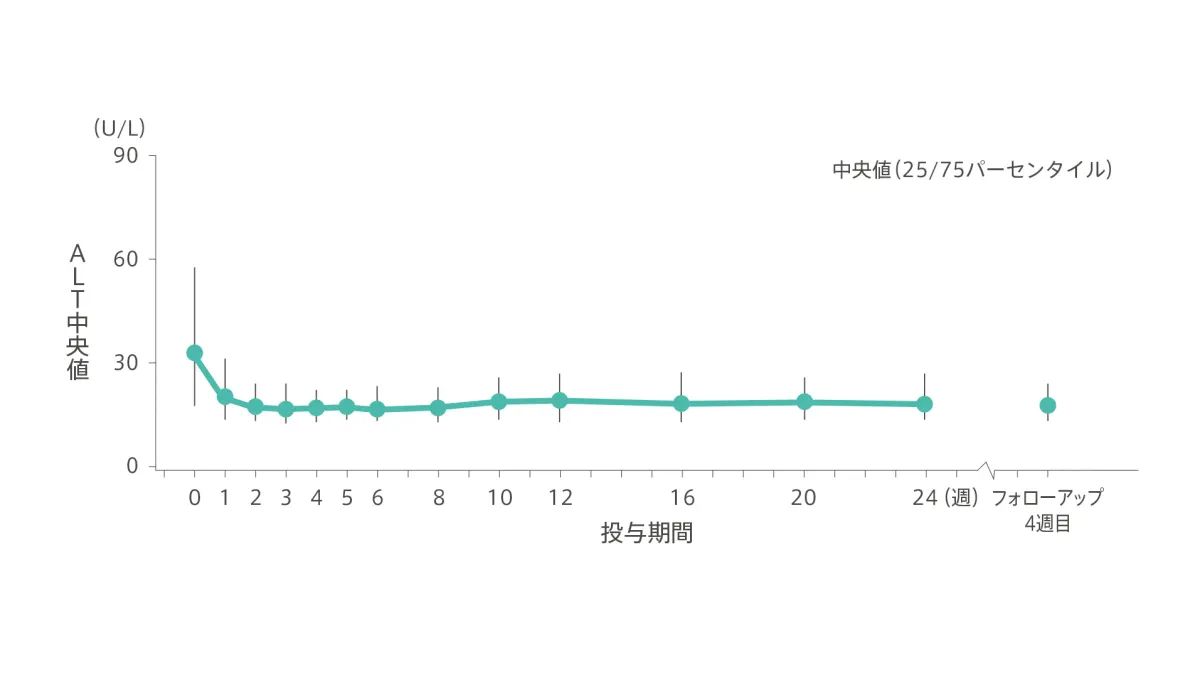
社内資料:承認時評価資料(国内第3相臨床試験:GS-US-342-3921)
Izumi N, et al. Hepatol Int. 12(4): 356-367, 2018
利益相反: 本研究はギリアド・サイエンシズ, Inc.の資金提供および支援により行われた。本論文の著者には、ギリアド・サイエンシズ, Inc.の社員が含まれる。本論文の著者には、ギリアド・サイエンシズ,
Inc.より講演料等を受領した者が含まれる。
副作用
前治療歴を有するC型慢性肝炎又はC型代償性肝硬変患者を対象に本剤とリバビリンを24週間併用投与した国内第3相臨床試験において、60例中21例(35.0%)に副作用が認められました。主な副作用は、貧血13例(21.7%)、倦怠感3例(5.0%)、そう痒症2例(3.3%)等でした。(承認時)
なお重大な副作用として、貧血[21.7%(前治療歴を有するC型慢性肝炎又はC型代償性肝硬変におけるウイルス血症の改善)]、高血圧[頻度不明※(効能・効果共通)]、脳血管障害[頻度不明※(効能・効果共通)]が報告されています。
- ソホスブビルを含有する製剤の製造販売後において報告されている副作用のため頻度不明。
◆ 直接作用型抗ウイルス薬(DAA)による治療歴を有するC型慢性肝炎又はC型代償性肝硬変患者における試験成績(国内第3相臨床試験)
| 安全性評価対象例数 | 60例 |
|---|---|
| 副作用発現症例数 | 21例 |
| 副作用発現症例率 | 35.0% |
| エプクルーサ配合錠+RBV 24週間投与群 |
|
|---|---|
| 副作用発現例数(%) | 21(35.0%) |
| 副作用の種類、例数(%) | |
| 血液及びリンパ系障害 | 13(21.7%) |
| 貧血 | 13(21.7%) |
| 内分泌障害 | 1(1.7%) |
| 甲状腺機能亢進症 | 1(1.7%) |
| 胃腸障害 | 2(3.3%) |
| 口内炎 | 1(1.7%) |
| 下痢 | 1(1.7%) |
| 一般・全身障害及び投与部位の状態 | 3(5.0%) |
| 倦怠感 | 3(5.0%) |
| 感染症及び寄生虫症 | 2(3.3%) |
| 咽頭炎 | 1(1.7%) |
| ウイルス性上気道感染 | 1(1.7%) |
| 代謝及び栄養障害 | 1(1.7%) |
| 食欲減退 | 1(1.7%) |
| 神経系障害 | 1(1.7%) |
| 頭痛 | 1(1.7%) |
| 精神障害 | 1(1.7%) |
| うつ病 | 1(1.7%) |
| 皮膚及び皮下組織障害 | 5(8.3%) |
| そう痒症 | 2(3.3%) |
| 発疹 | 1(1.7%) |
| 湿疹 | 1(1.7%) |
| 紅斑性皮疹 | 1(1.7%) |
MedDRA Version 20.0
承認時社内集計
その他関連製品


